Argenx Advances Clinical Development of ARGX-119 i...
Category : Clinical Trial Update 30 June 2025Overview argenx SE (Euronext & Nasdaq: ARGX), a global immunology company committed to improv...
Newsletter- All News is a comprehensive section of updated news and current trends, specifically curated to keep clinical trials, including breakthroughs in drug research, regulatory approvals, and advancements in study methodologies. Our news covers key trends in trial designs, patient recruitment strategies, and cutting-edge technologies shaping the future of clinical research
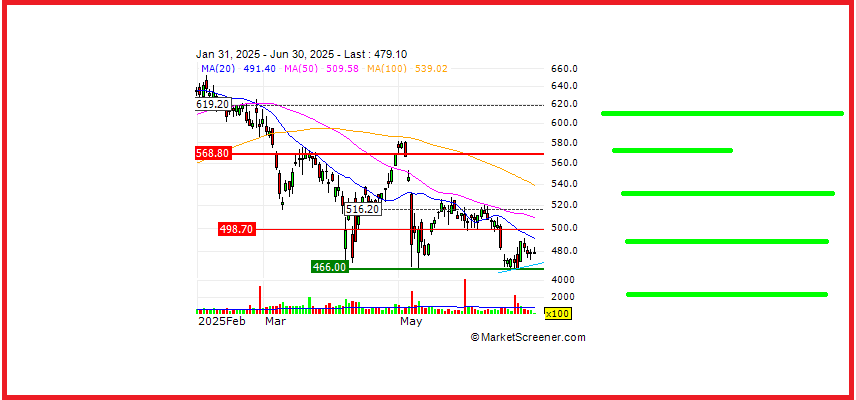
Overview argenx SE (Euronext & Nasdaq: ARGX), a global immunology company committed to improv...
Overview Bavarian Nordic A/S, a global vaccine company with a mission to improve health and save...

Overview UCB, a global biopharmaceutical company, today announced that the Phase 3 study investig...

Overview Nicox SA (Euronext Growth Paris: FR0013018124, ALCOX), an international ophthalmology co...

Overview Edgewise Therapeutics, Inc., a leading muscle disease biopharmaceutical company, unveile...
30 June 2025
30 June 2025
30 June 2025
30 June 2025

28 June 2025
C-89, Sector-65 Noida-U.P. 201301 (India)
Phone: +91-120-6631301-361
Mobile: +91-9990237670
Customer Care: 18004190155
Email: info@lifescienceintellipedia.com
Email: sales@lifescienceintellipedia.com